Global Humanized Mouse and Rat Model Market Latest Research Report (2023-2031) is segmented into Regions, Applications(Pharmaceutical, Biotechnology Companies), and Types(Genetic, others). The report presents the research and analysis provided within the Humanized Mouse and Rat Model Market Research is meant to benefit stakeholders, vendors, and other participants in the industry. This report is of 118Pages long. The Humanized Mouse and Rat Model Market is expected to grow annually by magnificent (CAGR 2023 2031).
Get a Sample PDF of report https://www.marketreportsworld.com/enquiry/request-sample/24317572 Transgenic And Gene Knockout Technology
Who is the largest manufacturers of Humanized Mouse and Rat Model Market worldwide?
Crown Bioscience (US) Horizon Discovery Group plc (UK) Vitalstar Biotechnology Co (China) AXENIS S.A.S (France) Harbour Antibodies BV (China) Hera BioLabs (US) The Jackson Laboratory (US) Hera BioLabs (US) Charles River Laboratories (US) Taconic Biosciences (US) Champions Oncology (US) inGenious Targeting Laboratory (US) Horizon Discovery Group plc (UK) genOway (France) Transgenic (Japan)
Get a Sample Copy of the Humanized Mouse and Rat Model Market Report 2023
Short Description About Humanized Mouse and Rat Model Market:
The Global Humanized Mouse and Rat Model Market is anticipated to rise at a considerable rate during the forecast period, between 2022 and 2031. In 2021, the market is growing at a steady rate and with the rising adoption of strategies by key players, the market is expected to rise over the projected horizon.
According to the latest research, the global Humanized Mouse and Rat Model market size was valued at USD million in 2022 and is expected to expand at a CAGR of Percent during the forecast period, reaching USD million by 2028.
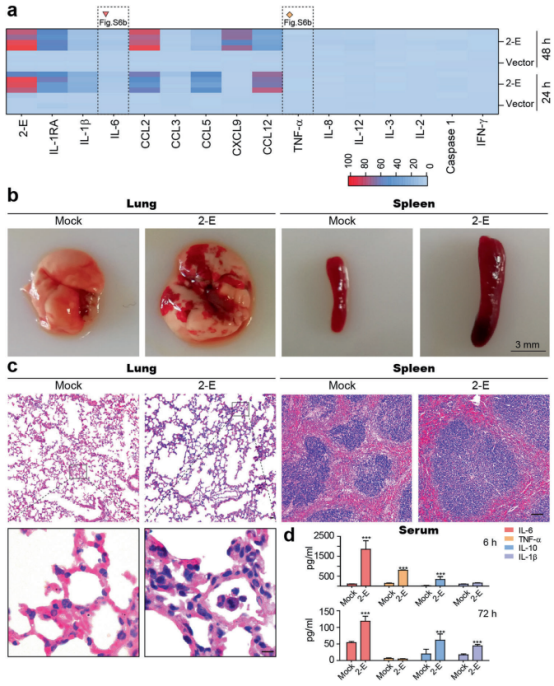
A "humanized" immune system rodent model is one in which human primary haematopoietic cells and tissues are implanted into an immunodeficient host that generate a functional human immune system. Humanized mice or rat model are commonly used as small animal models in biological and medical research for human therapeutics. Growth in this market can primarily be attributed to factors such as the growing demand of personalized medicine, continuous support in the form of investments and grants from the government and private sectors, increasing number of RandD activities in pharmaceutical and biotechnology companies, and the increasing number of research activities involving humanized models. Increasing production of monoclonal antibodies and rising demand for humanized rat models are expected to provide a wide range of growth opportunities for players in the market.
This report elaborates on the market size, market characteristics, and market growth of the Humanized Mouse and Rat Model industry between the year 2018 to 2028, and breaks down according to the product type, downstream application, and consumption area of Humanized Mouse and Rat Model. The report also introduces players in the industry from the perspective of the value chain and looks into the leading companies.
Ask for A Sample Report
What Are the Factors Driving applications of the Growth of the Humanized Mouse and Rat Model Market?
What Are the Types of Humanized Mouse and Rat Model Market Available in The Market?
The market is segmented based on the following product types, which in 2022 represented the largest share of the global Humanized Mouse and Rat Model market.
Inquire more and share questions if any before the purchase on this report at https://www.marketreportsworld.com/enquiry/request-sample/24317572
Which Regions Are Leading the Humanized Mouse and Rat Model Market?
To Understand How COVID-19 Impact is Covered in This Report. Request a Sample Copy of the Report
This Humanized Mouse and Rat Model Market Research/Analysis Report Contains Answers to your following Questions
Humanized Mouse and Rat Model Market Covid-19 Impact and Recovery Analysis:
We maintained updated on the immediate impact of COVID-19 in this market as well as its secondary impacts from many businesses. This article examines the pandemic's impact on the keyword market both globally and locally. According to kind, utility, and consumer sector, the study describes the market size, market characteristics, and market growth for the consumer goods contractual manufacturing business. Furthermore, it offers a comprehensive analysis of the additives involved in market development before and during the COVID-19 pandemic. Report further conducted a probing analysis of the industry to identify major influencers and entrance barriers. Our studies analysts will assist you to get custom designed info to your report, which may be changed in phrases of a particular region, utility or any statistical info. In addition, were constantly inclined to conform with the study, which triangulated together along with your very own statistics to make the marketplace studies extra complete for your perspective.
Purchase this report (Price PRPR USD for a single-user license) -https://www.marketreportsworld.com/purchase/24317572
Detailed TOC of Global Humanized Mouse and Rat Model Market Research Report 2023
1 Humanized Mouse and Rat Model Market Overview
1.1 Product Overview and Scope of Humanized Mouse and Rat Model
1.2 Humanized Mouse and Rat Model Segment by Type
1.3 Humanized Mouse and Rat Model Segment by Application
1.5 Global Market Size by Region
2.1 Global Humanized Mouse and Rat Model Production Capacity Market Share by Manufacturers (2017-2023)
2.2 Global Humanized Mouse and Rat Model Revenue Market Share by Manufacturers (2017-2023)
2.3 Humanized Mouse and Rat Model Market Share by Company Type (Tier 1, Tier 2 and Tier 3)
2.4 Global Humanized Mouse and Rat Model Average Price by Manufacturers (2017-2023)
2.5 Manufacturers Humanized Mouse and Rat Model Production Sites, Area Served, Product Types
2.6 Humanized Mouse and Rat Model Market Competitive Situation and Trends
3.1 Global Production Capacity of Humanized Mouse and Rat Model Market Share by Region (2017-2023)
3.2 Global Humanized Mouse and Rat Model Revenue Market Share by Region (2017-2023)
3.3 Global Humanized Mouse and Rat Model Production Capacity, Revenue, Price and Gross Margin (2017-2023)
3.4 North America Humanized Mouse and Rat Model Production
3.5 Europe Humanized Mouse and Rat Model Production
3.6 China Humanized Mouse and Rat Model Production
3.7 Japan Humanized Mouse and Rat Model Production
4 Global Humanized Mouse and Rat Model Consumption by Region
4.1 Global Humanized Mouse and Rat Model Consumption by Region
5.1 Global Humanized Mouse and Rat Model Production Market Share by Type (2017-2023)
5.2 Global Humanized Mouse and Rat Model Revenue Market Share by Type (2017-2023)
5.3 Global Humanized Mouse and Rat Model Price by Type (2017-2023)
6.1 Global Humanized Mouse and Rat Model Production Market Share by Application (2017-2023)
6.2 Global Humanized Mouse and Rat Model Revenue Market Share by Application (2017-2023)
6.3 Global Humanized Mouse and Rat Model Price by Application (2017-2023)
Get a Sample Copy of the Humanized Mouse and Rat Model Market Report 2023
Market Reports Worldis the Credible Source for Gaining the Market Reports that will Provide you with the Lead Your Business Needs. Market is changing rapidly with the ongoing expansion of the industry. Advancement in the technology has provided today's businesses with multifaceted advantages resulting in daily economic shifts. Thus, it is very important for a company to comprehend the patterns of the market movements in order to strategize better. An efficient strategy offers the companies with a head start in planning and an edge over the competitors.
Phone:US : +(1) 424 253 0946 UK : +(44) 203 239 8187 Email:sales@marketreportsworld.com Web:https://www.marketreportsworld.com
Find Out Our New Updated Reports Below :
Osteoarthritis Drugs Market Analysis Industry Dynamic Scenario | 2023-2030
Aircraft Seating Market Report Inside and Depth Analysis Growth and Emerging Trends |2030
Fitness Trackers Market Future Prospects, Business Growth and Development 2023|2030
Hair Color and Dye Market Research Report, Upcoming Business Opportunity, Expected Share Forecasted Period 2023|2030
Super Generics Market Research Report Demand and Future Trends Till 2030
Machine Vision Illumination Systems Market Segmentation Qualitative and Quantitative Research by 2023 - 2030
Mining Ball Mill Market Analysis Size Share and Growth Trends and Competitive Scenario Forecasted 2023-2030
Sustainable Aviation Fuel Market Share, Growth | Industry Report, 2023 -2030
Plastics Extrusion Market|2030 Industry Prospects Size Insights Growth
Digital Ad Fraud Detection Software Market Research Report Demand and Future Trends Till 2030
Fully Humanized Mouse © 2023 Benzinga.com. Benzinga does not provide investment advice. All rights reserved.