The number of items:0. Subtotal:$0.00
Shanghai Public Network Security No. 31011502016088 Shanghai ICP No. 08103356-1 Copyrights 2020 Accela ChemBio Inc. All Rights Reserved Hot Cas | Cas 9883 Pacific Heights Blvd., Suite H, San Diego, CA 92121 Tel: (858) 699-3322 Fax : (858) 876-1948 Inquiry/Orders: order@accelachem.com Sales Polymerization Inhibitor
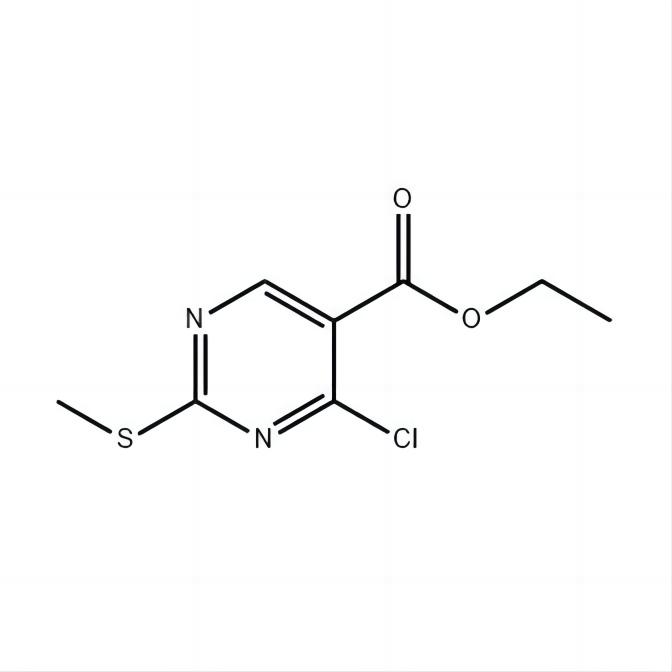
Shanghai (Pu) Emergency Management and Crisis Management Xu[2022]202788(YS)

Cytidine Monophosphate Disodium Uridine Dear valued customer, The product you ordered is out of stock. Our sales representatives will contact you for the lead time soon later. Thank you for your understanding!